Neurofibromatosi cutanea è il termine che identifica le “Neurocristopatie”, patologie ereditarie e secondarie a una displasia della cresta neurale.
INDICE ARTICOLO
Dal punto di vista clinico, fisiopatologico e molecolare, sotto questo nome vengono raggruppate malattie eterogenee, tra le quali ricordiamo le Neurofibromatosi, la Sclerosi tuberosa di Bourneville, le sindromi da neoplasie endocrine multiple e le facomatosi pigmentovascolari.
Neurofibromatosi cutanea: cosa sono
Le neurofibromatosi sono patologie ben distinte tra loro, ma accomunate da alcune manifestazioni cutanee che hanno la stessa origine embrionale. Si riconoscono almeno due malattie differenti, la cui modalità di trasmissione è autosomica dominante: la Neurofibromatosi di tipo 1 o malattia di von Recklinghausen (NF-1) e la Neurofibromatosi di tipo 2 (NF-2).
Classificazione delle Neurofibromatosi
Si utilizza la classificazione di Riccardi, basata su criteri clinici, per distinguere le varie tipologie di Neurofibromatosi dei bambini.
Nello specifico, NF-1 (Neurofibromatosi 1) e NF-2 (Neurofibromatosi tipo 2) hanno criteri diagnostici ben codificati, motivo per il quale sono quelle più chiaramente distinguibili.
Esiste anche una Neurofibromatosi di tipo 3 (NF-3) che viene definita come una neurofibromatosi mista poichè è caratterizzata da alcune manifestazioni tipiche sia della NF-1 sia della NF-2.
Esistono poi altre tipologie di Neurofibromatosi quali:
- NF-4 che comprende le forme variabili di Neurofibromatosi, le quali si differenziano sia sul piano clinico sia su quello genetico.
- NF-5 definita neurofibromatosi segmentale.
- NF-6 che si manifesta unicamente con numerose macchie caffelatte.
- NF-7 che include tutte le forme di NF-1 a esordio tardivo, che si manifestano dopo i 30 anni.
In ultimo, la NF-8 viene indicata come “di tipo non specificato” perché raggruppa tutte le forme incertae sedis che presentano aspetti tipici di una neurofibromatosi.
Neurofibromatosi di tipo 1 (Malattia di von Recklinghausen)
E’ la forma di Neurofibromatosi più frequente, con una prevalenza di circa 1/3000-1/3500 nati, e si trasmette con modalità autosomica dominante.
Il gene della NF-1 è stato localizzato sul cromosoma 1, in posizione 17q11.2.
La sua penetranza è virtualmente pari al 100% all’età di 5 anni, e le mutazioni de novo si manifestano in circa la metà dei casi.
La neurofibromina, prodotto del gene NF-1, è una proteina che interviene nel controllo del differenziamento e della proliferazione cellulare, poiché coinvolta nella regolazione di p21-ras, il prodotto del proto-oncogene c-ras.
Neurofibromatosi cutanea diagnosi
Criteri diagnostici per NF-1 (Neurofibromatosi tipo 1)
Per fare diagnosi di NF-1 il quadro clinico del paziente deve soddisfare almeno due dei sette criteri diagnostici indicati di seguito, tre dei quali sono dermatologici.
Neurofibromatosi cutanea sintomi
- Presenza di almeno sei macchie caffelatte, il cui diametro maggiore sia superiore a 5 mm se il soggetto è in età prepubere, e a 5 mm dopo la pubertà.
- Presenza di almeno due neurofibromi di qualsiasi tipo o di un neurofibroma plessiforme.
- Presenza di lentiggini ascellari o inguinali.
- Presenza di un glioma del nervo ottico.
- Presenza di almeno due noduli di Lisch (amartomi dell’iride).
- Presenza di una lesione ossea caratteristica come una displasia dello sfenoide, o un assottigliamento della corticale delle ossa lunghe, con o senza pseudoartrosi.
- Un parente di primo grado affetto da NF-1 diagnosticata in base ai criteri precedenti.
Macchie caffe latte: come sono e come fare per riconoscerle
Le macchie caffè latte, spesso congenite, raramente compaiono dopo i 2 anni e rappresentano la prima manifestazione della NF-1.

Presentano margini netti e un colore più o meno bruno, a volte al limite della visibilità. Si distribuiscono in modo casuale. Il diametro varia da 0,5 a 50 cm, ma nella maggior parte dei casi non supera 10 cm. In età adulta, queste macchie sono presenti in oltre il 90% dei casi.
Lentiggini
Compaiono dopo le macchie caffelatte e sono simili a queste, ma di diametro inferiore (1-3 mm). Raramente si riscontrano prima dei 2 anni di età e sono presenti in circa l’80% dei casi in età adulta. E’ di grande importanza diagnostica la presenza di lentiggini ascellari multiple, identificata come segno di Crowe.
Neurofibromi cutanei
Si distinguono tre tipologie di neurofibromi cutanei:
- dermici
- nodulari periferici , isolati o “a grappolo”
- plessiformi
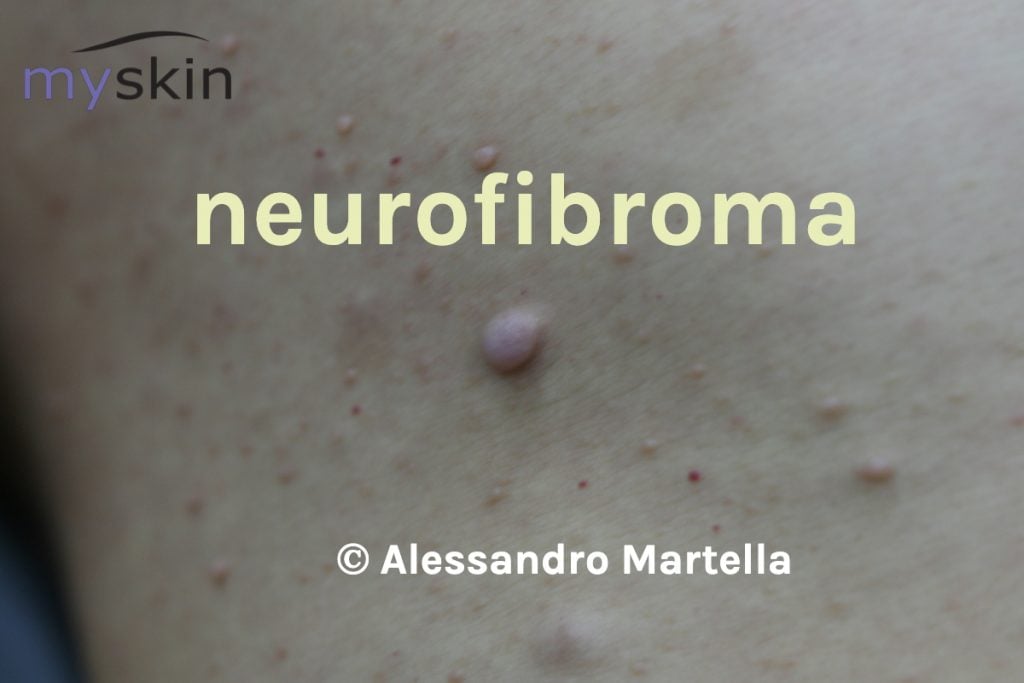
I primi sono piccole manifestazioni di consistenza molle, mobili alla palpazione, di colore rosa o violaceo, che si sviluppano nel derma. Hanno dimensioni che variano da 0,1 cm ad alcuni cm.
In alcuni casi si manifestano poche lesioni, in altri possono raggiungere qualche migliaio. Generalmente interessano il tronco e possono somigliare a fibromi penduli, ma anche altre aree del corpo ne possono essere affette. I neurofibromi dermici in un quarto dei casi possono essere pruriginosi e dolorosi. Hanno esordio nella pubertà e sono presenti in circa il 95% dei pazienti adulti.
I neurofibromi nodulari periferici sono presenti solo nel 15% dei pazienti affetti da NF-1. Sono di consistenza dura e interessano principalmente i tronchi nervosi. Se vengono compressi, possono causare parestesie.
Possono determinare una neuropatia sensitiva o motoria. Dal punto di vista clinico e istologico si presentano come tumefazioni sottocutanee che possono avere dimensioni variabili da alcuni centimetri a un intero segmento corporeo. La cute che li riveste si presenta anormale, con un mix di ipertricosi, ipertrofia e pigmentazione caffelatte.
Un neurofibroma plessiforme è presente fin dal primo anno di vita in almeno un terzo dei pazienti affetti da NF-1. Questi tumori colpiscono principalmente il tronco, ma possono interessare anche la testa, gli arti e il collo. Prendono il nome di “tumori regali” i grandi neurofibromi plessiformi dermatocalasici.
Noduli di Lisch
Si tratta di formazioni tumorali benigne (amartomi) dell’iride, che non causano problemi di visione alla persona. Sono manifestazioni difficilmente o per nulla osservabili ad occhio nudo ma che invece possono essere messi in evidenza con l’esame ottico della lampada a fessura. Col progredire dell’età, aumentano sia le loro dimensioni sia il loro numero.
Si riscontrano già nel 10% dei pazienti sotto i 6 anni e in più del 90% dei casi di pazienti sopra i 16 anni.
Glioma del nervo ottico
E’ presente in circa il 15% dei pazienti affetti da NF-1 e si rileva attraverso l’utilizzo di tecniche di imaging cerebrale.
Nell’1% dei casi, questo tumore può causare un idrocefalo, un esoftalmo, turbe ipotalamo-ipofisarie e una pubertà precoce, e può compromettere la prognosi quoad visum.
Evolve entro i primi 6 anni di vita. Dal punto di vista istologico, i gliomi del nervo ottico sono assimilabili ad astrocitomi pilocitici a basso grado di malignità. In alcuni casi siamo in presenza di lesioni gliotiche non tumorali dei nervi ottici difficili da distinguere dai gliomi veri e il cui trattamento è controverso.
Manifestazioni ossee
Le displasie delle ossa lunghe sono di natura congenita. Dal punto di vista clinico possono manifestarsi precocemente (curvatura congenita di una gamba) o apparire al momento in cui il soggetto inizia a deambulare, come nel caso delle fratture con pseudoartrosi secondarie che colpiscono dall’1 al 3% dei pazienti.
Complicanze della NF-1
Le complicanze più frequentemente associate a NF-1 sono dovute ai neurofibromi plessiformi che, localizzati a livello cerebrale, possono causare difficoltà di apprendimento che si riscontrano nel 30-40% dei casi.
Solo nell’1-2% dei casi si ritrovano altre complicanze, considerate più rare: l’idrocefalo, le pseudoartrosi, i gliomi ottici aggressivi, i glioblastomi cerebrali (astrocitomi ad alto grado di malignità), le scoliosi bisognose di correzione chirurgica, i feocromocitomi, i carcinoidi, i neurofibromi nodulari profondi con tendenze compressive e i neurofibrosarcomi.
Una grave complicazione si presenta con una frequenza del 5% nei pazienti adolescenti e adulti ed è caratterizzata dal neurofibrosarcoma o tumore maligno delle guaine nervose. La degenerazione neoplastica di solito interessa i neurofibromi nodulari periferici o plessiformi.
Solo in casi eccezionali si riscontra associazione tra xantogranuloma giovanile e leucemia mielomonocitaria.
Gestione dei pazienti con NF-1
Se i neurofifromi osservabili ad occhio nudo, le macchie caffè latte, le lentiggini possono rappresentare solo un problema estetico, quello che è importante di fronte ad una persona con NF-1 il follow-up clinico per individuare in modo tempestivo le eventuali complicanze descritte sopra.
Proprio per questo è consigliabile la gestione multidisciplinare della persona con NF-1 a causa della differente e varia manifestazione clinica che può interessare la cute ma anche gli organi interni.
Ad esempio, in presenza di un glioma si consiglia l’imaging dei nervi ottici e del chiasma mediante RMN il suo monitoraggio nel corso della vita.
Neurofibromatosi cura
Ad oggi non esistono ancora trattamenti specifici per NF-1 e non si può prevedere il decorso della malattia in un dato paziente.
Invece i neurofibromi cutanei possono essere trattai sia con diverse tecniche chirurgiche, quali l’asportazione oppure il trattamento laser CO2.
Le altre Neurofibromatosi
Neurofibromatosi di tipo 2
Questa Neurofibromatosi è molto rara e si manifesta in un caso su 33.000-40.000.
Il gene della Neurofibromatosi di tipo 2 si trova sul cromosoma 22 in posizione 22q12.2.
Nella NF-2 sono presenti Schwannomi vestibolari bilaterali, Schwannomi di altri nervi cranici e nervi spinali, e meningiomi. Le lesioni cutanee non sono frequenti, meno del 10% dei pazienti affetti da NF-2 manifesta più di due macchie caffelatte.
Per fare diagnosi di NF-2 è necessario rilevare una delle seguenti evidenze:
- presenza di Schwannomi bilaterali dell’VIII paio dei nervi cranici
- presenza di un Schwannoma unilaterale del nervo stato-acustico (vestibolococleare) o di due manifestazioni tra: neurofibroma, meningioma, ependimoma, schwannoma in altra sede, cataratta posteriore giovanile.
Neurofibromatosi segmentale o NF-5
La NF-5 si manifesta solo in casi eccezionali, con una prevalenza inferiore allo 0,001%.
I segni che la identificano sono macchie caffelatte, neurofibromi, lentiggini a livello di un solo segmento corporeo, emisoma. Più rara è la presenza di noduli di Lisch o di diversi segmenti bilateralmente.
Si può fare diagnosi di Neurofibromatosi segmentaria solo nel caso in cui si esclude la NF-1 e la NF-2.
Si pensa che questa tipologia di neurofibromatosi derivi da una mutazione somatica post-zigotica.
Altre forme rare di Neurofibromatosi
- NF-1 associata alla sindrome di Noonan: collo corto con pterygium colli, ptosi palpebrale, piccola taglia corporea, pectus excavatum, impianto basso delle orecchie e malformazioni cardiache
- NF-3: presenza contemporanea di aspetti tipici di NF-1 e NF-2
- NF-7: neurofibromatosi con insorgenza dei neurofibromi dopo i 30 anni
casi di neurofibromatosi con presenza esclusiva di anomalie ossee o macchie caffelatte
sindrome di Watson: malattia autosomica dominante che si identifica per l’associazione ritardo mentale, macchie caffelatte e restringimento patologico dell’arteria polmonare
Diagnosi differenziale delle Neurofibromatosi
Sindrome di Proteo
E’ una malattia rara a lungo confusa con la NF-1. Si caratterizza per l’associazione variabile di un’emi-ipertrofia di uno o più segmenti corporei, di una macrodattilia, di nevi connettivali e/o epidermici.
Costante è l’ispessimento delle palme e delle piante che assumono un aspetto cerebriforme. Istologicamente, gli ispessimenti sottocutanei si caratterizzano come tumori angiomatosi, lipomatosi o amartomatosi.
Le Neurofibromatosi rappresentano un gruppo così eterogeneo di patologie che devono essere distinte anche da:
- le melanosi cutanee
- la sindrome di Klippel-Trénaunay (angioma ed emi-ipertrofia)
- la MEN2B
- la sindrome di von Hippel-Lindau
- la sindrome di Carney
- le lipomatosi
Infine, forme affini alla NF-2 sono le Schwannomatosi o Neurilemmatosi che si differenziano dalla NF-2 per la presenza di schwannomi multipli ma in assenza di Neurinomi dell’acustico.
Risorsa Utile:
ANF Associazione per la Neurofibromatosi onlus
Bibliografia
- Neurofibromatosis. Korf BR. Handb Clin Neurol. 2013;111:333-40
- Neurofibroma. Messersmith L, Krauland K

