Il pemfigoide bolloso rientra nel gruppo delle dermatosi bollose autoimmuni sottoepidermiche, delle quali ne rappresenta un buon 70%.
Colpisce senza distinzione di sesso e razza gli adulti sopra i 70 anni, solo in modo eccezionale i bambini.
INDICE ARTICOLO
Aspetto clinico
I primi sintomi di questa patologia sono caratterizzati da prurito in assenza di manifestazioni cliniche, sonno disturbante e cronico, “intertrigini” inguinali o sottomammarie e sviluppo di chiazze simili a eczema o orticaria.
L’eruzione cutanea caratteristica del pemfigoide bolloso si presenta sotto forma di bolle tese che insorgono spontaneamente, Nikolski-negative, con all’interno un liquido limpido. Queste lesioni vanno da 0,5 a svariati centimetri e si sviluppano a ridosso di chiazze eritematose. Alle bolle si associano anche macule e papule simili a eritemi o orticaria, e croste ed erosioni causate dalla rottura delle bolle stesse.
Tutte queste lesioni si associano a un intenso prurito.
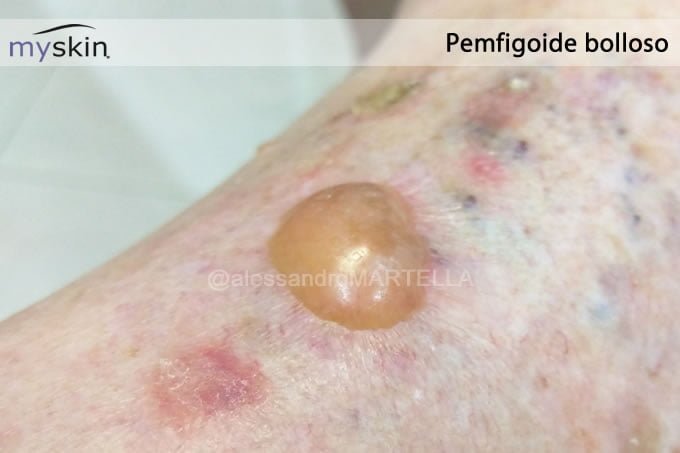
Le manifestazioni cutanee di questa patologia si sviluppano in modo simmetrico nelle regioni flessorie degli arti, nella zona anteriore delle cosce e sull’addome. Solo nel 10-20% dei casi ne è affetta anche la mucosa orale.
Alla guarigione, non restano grani di miglio o cicatrici distrofiche.
Esistono numerose forme atipiche di questa patologia come:
- il pemfigoide “disidrosiforme”, caratterizzato dallo sviluppo di bolle nelle sedi palmoplantari o su cicatrici
- lesioni vescicolari simili a eczema erpetiforme
- lesioni delle grandi pieghe o del tipo di una prurigo nodulare
- eritrodermia in casi eccezionali.
Decorso
Questa patologia può evolvere in modo imprevedibile, motivo per il quale non è possibile esprimersi sulla prognosi. Nei pazienti anziani sono frequenti e spesso molto gravi gli effetti collaterali dovuti alla terapia a base di corticosteroidi, così come lo sviluppo di resistenze a questi farmaci o dipendenza da essi. Frequenti sono le recidive a distanza di 2-6 anni dalla sospensione del trattamento. Il tasso di mortalità a distanza di un anno dalla diagnosi si aggira tra il 12 e il 30% dei casi.
Associazioni
Il pemfigoide bolloso solo raramente (o casualmente) si associa a malattie come artrite reumatoide, vitiligine, lupus eritematoso, pemfigo volgare, per citarne alcuni.
Al contrario, chi soffre di questa patologia ha un rischio maggiore di sviluppare anche psoriasi, sclerosi multipla e diabete
Diagnosi
E’ stato dimostrato che i pazienti affetti da pemfigoide bolloso presentano di frequente ipereosinofilia, a volte marcata, che si può associare a un innalzamento delle IgE sieriche.
Istologia
Dall’esame istologico effettuato su un campione prelevato da una bolla cutanea di recente formazione si possono individuare aspetti di bolla sottoepidermica, senza necrosi del tetto né acantolisi, nella quale sono presenti fibrina ed elementi cellulari come olimorfonucleati neutrofili ed eosinofili.
Studi immunopatologici
Il metodo dell’immunofluorescenza diretta ha un alto valore diagnostico se utilizzato su un campione di cute peribollosa prelevato tramite biopsia in quanto permette di individuare depositi lineari di IgG e/o di C3 (a volte associati ad altre immunoglobuline) lungo la membrana basale dell’epidermide.
Applicando l’immunofluorescenza indiretta su un campione di cute intatto prelevato da un paziente affetto da pemfigoide si può rilevare la presenza di IgG sierici anti-membrana basale dell’epidermide in circa il 90% dei casi.
L’immunoprecipitazione e l’immunostampa rilevano la presenza di autoanticorpi sierici specifici in circa l’80% dei pazienti affetti dal pemfigoide bolloso.
Attraverso l’immunomicroscopia elettronica diretta su campioni prelevati da cute peribollosa è possibile evidenziare depositi di immunoreattanti (IgG, C3) situati nella parte alta della lamina lucida, con rafforzamento del segnale in corrispondenza degli emidesmosomi.
Diagnosi differenziale
L’immunofluorescenza diretta e l’esame istologico convenzionale permettono di differenziare il pemfigoide bolloso da pemfigo, dermatite erpetiforme, eritema polimorfo, dermatosi a IgA lineari e tossidermie bollose.
L’immunofluorescenza indiretta su cute separata consente di distinguere il pemfigoide bolloso da una forma infiammatoria dell’epidermolisi bollosa acquisita.
Esami immunopatologici più raffinati, come l’immunomicroscopia elettronica e il Western blot, permettono di escludere le dermatosi bollose autoimmuni sottoepidermiche a depositi lineari di IgG e/o C3 in tutti i casi atipici con prevalente interessamento delle mucose, quando sono colpite sedi insolite, o quando le lesioni evolvono lasciando esiti cicatriziali.
Patogenesi
E’ stato ormai ampiamente dimostrato che il bersaglio degli autoanticorpi (BP230 e BP180) nel pemfigoide bolloso sono gli emidesmosomi, ossia quelle strutture che connettono l’epidermide alla lamina lucida favorendo la coesione dermo-epidermica.
Trattamento del Pemfigoide bolloso
Fino alla completa riepitelizzazione delle erosioni lasciate dalle bolle, il trattamento viene eseguito in ospedale, ogni giorno, per evitare sovrainfezioni locali o sistemiche.
Vengono praticati bagni con asettici e successivamente si applicano topici oleosi o sulfadiazina argentica. La medicazione si completa con tulle grasso sterile.
Forme estese ed evolutive di pemfigoide bolloso vengono trattate in via preferenziale in modo sistemico, con prednisone o prednisolone (1 mg/kg/die), facendo attenzione a compensare carenze vitaminiche, perdite idriche e proteiche.
Nei casi in cui si manifesta corticoresistenza, corticodipendenza o controindicazione al trattamento, si può optare per terapie alternative, come ad esempio l’associazione di immunosoppressori (azatioprina, 100-10 mg/die, o clorambucil, 4-6 mg/die) ai cortisonici.
Corticosteroidi topici di classe I vengono prescrizzi a quei pazienti che presentano forme di pemfigoide bolloso localizzate, paucilesionali e/o scarsamente evolutive.

